引言
Introduction
AI 辅助酶改造技术再现突破,我公司创始人兼 CTO,上海交通大学生命科学技术学院杨广宇教授团队,联合上海交通大学自然科学研究院洪亮教授团队,应用 Pro-PRIME 蛋白语言大模型和高效模型定向精调,仅用两轮,即实现蛋白稳定性大幅提升,且复合突变 100% 增益成功。

杨广宇 教授
瀚海新酶 CTO
该技术基于蛋白质大模型,仅用有限的目标酶蛋白的(突变-活性-稳定性)标注数据集(<100),通过模型精调,准确预测复合突变对目标酶蛋白的稳定性影响,助力酶稳定性改造过程中的序列设计。相比单纯的高通量筛选方法,具有效率高,成本低,工作量少,筛选容易的优点,为基于 AI 技术进行酶改造提供了新的思路和方向。

Overview
概述
优化酶的热稳定性对于蛋白质科学和工业应用至关重要。目前,通过(半)理性设计和随机诱变方法可以较为准确地设计多个增强酶热稳定性的单点突变。但当组合多个突变时,常常会出现复杂的上位效应,导致组合突变体完全失活。因此,优化酶通常需要进行多轮设计,从而逐步引入单个突变位点,整个过程非常耗时。
近日,我公司创始人兼 CTO,上海交通大学生命科学技术学院杨广宇教授团队的文章 “Optimizing enzyme thermostability by combining multiple mutations using protein language model” 在 《mLife》正式上线,上海交通大学自然科学研究院洪亮教授为共通讯。该研究团队提出了一种人工智能辅助的酶热稳定性工程策略,可以高效地组合多个有益单点突变。在肌酸酶的进化实例中,仅经过两轮设计,获得了 50 个具有卓越热稳定性的组合突变体,设计成功率达 100%。经少量实验数据微调后的模型可以从数据集中有效捕捉组合突变体中的上位效应。
Abstract
摘要原文
文末阅读原文章
“Optimizing enzyme thermostability is essential for advancements in protein science and industrial applications. Currently, (semi-)rational design and random mutagenesis methods can accurately identify single-point mutations that enhance enzyme thermostability. However, complex epistatic interactions often arise when multiple mutation sites are combined, leading to the complete inactivation of combinatorial mutants. As a result, constructing an optimized enzyme often requires repeated rounds of design to incrementally incorporate single mutation sites, which is highly time-consuming. In this study, we developed an AI-aided strategy for enzyme thermostability engineering that efficiently facilitates the recombination of beneficial single-point mutations. We utilized thermostability data from creatinase, including 18 single-point mutants, 22 double-point mutants, 21 triple-point mutants, and 12 quadruple-point mutants. Using these data as inputs, we used a temperature-guided protein language model, Pro-PRIME, to learn epistatic features and design combinatorial mutants. After two rounds of design, we obtained 50 combinatorial mutants with superior thermostability, achieving a success rate of 100%. The best mutant, 13M4, contained 13 mutation sites and maintained nearly full catalytic activity compared to the wild-type. It showed a 10.19°C increase in the melting temperature and an ~655-fold increase in the half-life at 58°C. Additionally, the model successfully captured epistasis in high-order combinatorial mutants, including sign epistasis (K351E) and synergistic epistasis (D17V/I149V). We elucidated the mechanism of long-range epistasis in detail using a dynamics cross-correlation matrix method. Our work provides an efficient framework for designing enzyme thermostability and studying high-order epistatic effects in protein-directed evolution.”
Main content
主要内容
在该项研究中,作者利用一种 AI 辅助的酶热稳定性工程策略,通过少量实验数据微调 Pro-PRIME 模型来预测组合突变体的稳定性和活性。其中,Pro-PRIME 模型是基于 9600 万个宿主细菌菌株的最佳生长温度数据进行训练的蛋白质语言模型,在设计和优化高温酶方面表现优异。微调所使用的初始数据集包括来自肌酸酶的 73 个低阶突变体的序列-热稳定性和活性数据。然后使用微调后的模型来预测来自 18 个单点突变体的所有可能突变体的热稳定性和活性。主要目标是在保持至少 60% 的相对活性(相对于野生型),同时增强热稳定性的突变体(图 1)。

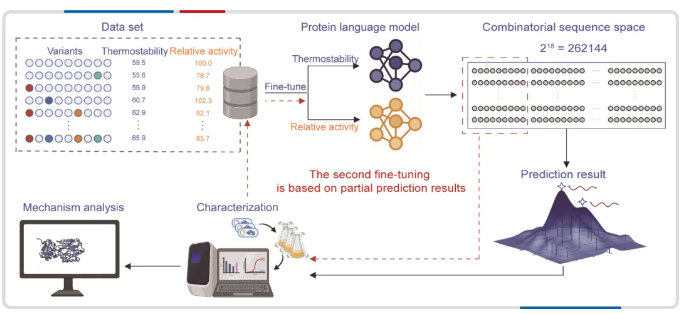
图1. 基于蛋白质语言模型组合突变的策略。
整个过程包括四个步骤:(1) 收集数据,(2) 蛋白质语言模型的微调,(3) 在组合序列空间中预测所有突变体,以及 (4) 验证所选突变体。红色虚线是第二轮模型微调。
为了进一步提高预测精度,研究人员将第一轮预测的实验表征结果整合到数据集中,并进行了第二轮微调、预测和选择。两轮微调和预测过程仅用了两周时间,共设计 50 个组合突变体,实现了 100% 的热稳定性设计成功率(图 2)。
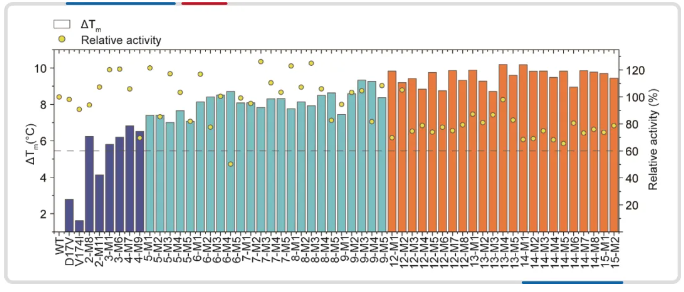
图2. 组合突变体的热稳定性和相对活性数据。
黄色圆圈是指:相对活性数据。条形图是指:突变体的热稳定性数据,其中蓝色、青色和橙色分别指初始数据集、第一轮和第二轮预测的数据集。
其中,最佳突变体 13M4 包含 13 个突变位点,与野生型相比,它的活性基本保持不变,在 Tm 上提高了 10.19°C,在 58°C 下的半衰期增加了约 655 倍。
在回顾数据时发现,即使某些突变在空间上相距甚远,也存在复杂的高阶上位性效应。例如 K351E 单点突变表现为阴性突变,但其在高阶突变体中却表现为阳性突变。此外,单点突变 D17V 和 I149V 存在明显的协同作用。结果表明,使用高质量的实验数据微调模型的参数,可以帮助模型准确捕获数据集中的已存在的上位效应,并用于后续高阶组合突变体的适应度预测。
动态相关矩阵分析的结果表明,影响稳定性的突变不仅影响其局部环境的动力学,在某些情况下,还影响远端结构区域的动力学(图 3)。该项技术可以作为未来研究或设计上位效应的一个有效工具。
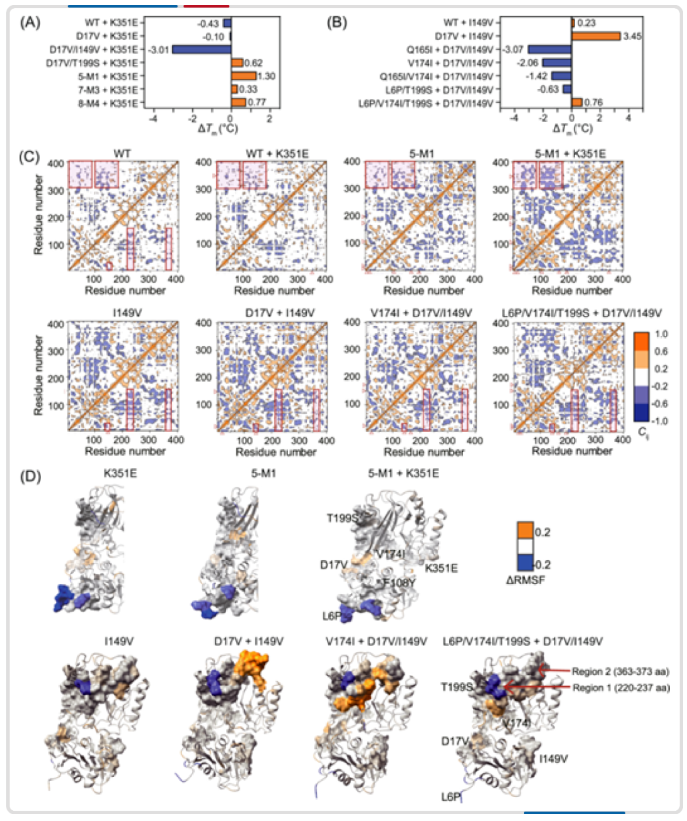
图3. 突变之间的上位效应分析。
K351E (A) 和 D17V/I149V (B) 在 Tm 值上的上位效应。蓝色表示负效应,橙色表示正效应。(C) 肌酸酶野生型和对应突变体的动态互相关矩阵图。相关系数 (Cij) 用不同颜色表示。突变位点用红色箭头标出,突变周围显著的动力学相关区域用红色框突出显示。(D) 突变体结构与野生型结构比较的的标准化 RMSF 变化。
Key Highlights
主要亮点
PART.01
本研究提出的人工智能辅助的酶热稳定性工程策略,可以高效地组合多个有益单点突变。仅通过两轮设计,共表征 50 个组合突变体,稳定性设计成功率达到 100%。与野生型相比,最佳突变体 13M4 的 Tm 提升了 10.19℃,58℃ 半衰期增加 655 倍,同时催化活性保持不变。
PART.02
通过利用少量但高质量的实验数据微调蛋白质语言模型参数,微调后的模型可以准确捕捉初始数据集中的上位效应,包括符号和协同上位效应。这表面,实验数据对于提升模型对高阶组合突变体的预测性能至关重要。
PART.03
利用动态相关矩阵分析,该研究揭示了长程上位效应的机制,显示了远距离突变之间在动力学上的相关性,从而共同影响突变体的稳定性。
PART.04
通过采用这种策略,研究团队仅通过两轮设计,全面探索了组合序列空间中超 26 万种可能的突变体,最佳突变体包含 13 个突变,大大缩短了传统方法中所需要的进化轮次,提升了蛋白质工程的效率。
PART.05
研究强调了将来自蛋白质工程的数据与先进的人工智能模型相结合,可以进一步提升模型的预测性能,从而提升蛋白质工程效率。该策略可以推广应用至多种关键酶分子的进化任务中。
识别二维码,立即浏览原文

欢迎与我们联系!

内容丨研发中心
编辑排版丨品牌部
审核丨研发中心、品牌部
图片来源丨瀚海新酶